Prolific researcher Chiara Barbieri has put online another interesting study on African genetics, this time about the Bantu populations of Southwestern and Central-Southern Africa (i.e. Namibia, Angola, Botswana and Zambia).
Chiara Barbieri et al., Migration and interaction in a contact zone: mtDNA variation among Bantu-speakers in southern Africa. bioRXiv 2014. Freely accessible (pre-pub) → LINK
ABSTRACT
Bantu speech communities expanded over large parts of sub-Saharan Africa within the last 4000-5000 years, reaching different parts of southern Africa 1200-2000 years ago. The Bantu languages subdivide in several major branches, with languages belonging to the Eastern and Western Bantu branches spreading over large parts of Central, Eastern, and Southern Africa. There is still debate whether this linguistic divide is correlated with a genetic distinction between Eastern and Western Bantu speakers. During their expansion, Bantu speakers would have come into contact with diverse local populations, such as the Khoisan hunter-gatherers and pastoralists of southern Africa, with whom they may have intermarried. In this study, we analyze complete mtDNA genome sequences from over 900 Bantu-speaking individuals from Angola, Zambia, Namibia and Botswana to investigate the demographic processes at play during the last stages of the Bantu expansion. Our results show that most of these Bantu-speaking populations are genetically very homogenous, with no genetic division between speakers of Eastern and Western Bantu languages. Most of the mtDNA diversity in our dataset is due to different degrees of admixture with autochthonous populations. Only the pastoralist Himba and Herero stand out due to high frequencies of particular L3f and L3d lineages; the latter are also found in the neighboring Damara, who speak a Khoisan language and were foragers and small-stock herders. In contrast, the close cultural and linguistic relatives of the Herero and Himba, the Kuvale, are genetically similar to other Bantu-speakers. Nevertheless, as demonstrated by resampling tests, the genetic divergence of Herero, Himba, and Kuvale is compatible with a common shared ancestry with high levels of drift and differential female admixture with local pre-Bantu populations.
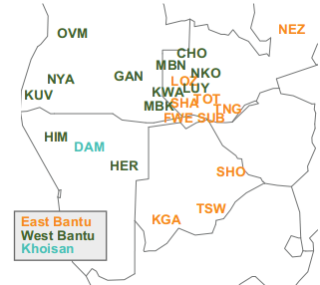 |
|
Figure 1: Map showing the rough geographical location of populations,
colored by linguistic affiliation. Abbreviations of population labels are
as specified in Table 1.
|
In spite of the Bantu-centric approach of the study, which also has its merits, my greatest interest is rather in the less typically Bantu lineages, which speak of admixture with several pre-Bantu populations.
In this sense I find the following highlights:
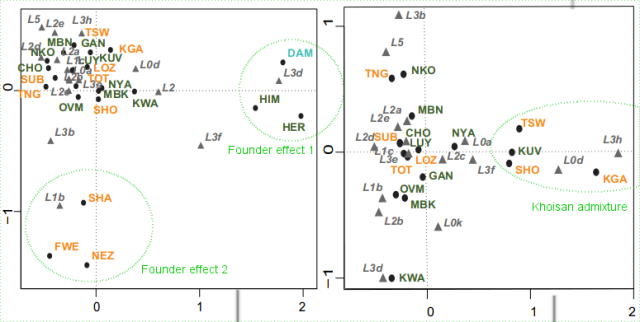 |
| Fig. S2 (annotated in green by Maju): CA plots based on haplogroup frequencies. Left: all the dataset, right: excluding outliers. |
L3d and L3f founder effect:
The Himba and Herero, as well as the non-Bantu pastoralists Damara make one distinctive cluster defined by the high frequencies of haplogroup L3d, as well as L3f (not present among the Damara but found among the Kuvale). As discussed in the paper, the Himba and Herero may be related to the Kuvale of SW Angola but they have notable differential levels (or directionality) of aboriginal admixture.
As both L3d and L3f are present in West and East Africa alike, it is interesting to track the specific subhaplogroups implicated in this founder effect, something done in fig. 4.
The main L3d sublineage is
L3d3a1, whose haplotype network shows a largely Khoisan centrality (not Damara) although this node is shared also by some unspecified “other Bantu”. The Southern Africa specificity of L3d3a was already noticed in the past (see
here). So it is very possible that we are before an aboriginal Southern African lineage, maybe arrived with the first Khoisan Neolithic (or whatever other ancient flow) rather than a Bantu-specific founder effect.
The main L3f subhaplogroup is
L3f1b4a, which seems more specifically Bantu, with a major branch concentrated among the Himba, Herero and Kuvale. This lineage is not found among the Damara in spite of the other strong affinity of this Khoisan population towards the Himba and Herero. L3f1b is found in Southern Africa, Kenya and Oman (
per Bihar 2008), so we are probably before a distinctive East African element, not too likely to be genuinely Bantu but possibly just assimilated into Bantu ethnic identity.
Even if both lineages converge in the Himba and Herero, they are almost certainly different inputs, one of Damara (herder Khoisan) origin and the other of Bantuized East African origin maybe.
L1b founder effect:
L1b is essentially a West African lineage
concentrated in the Sahel area from Chad westwards (although L1b1a2 is from the Nile basin). A particularly high frequency population are the Fulani pastoralists, original from the Westernmost African plateaus, who ruled many kingdoms in West Africa between the collapse of the colonial rule by Morocco and the consolidation of the European conquest of the continent.
As this study does not dwell in sublineages, we cannot understand the most likely specific origins of it among several Southern African populations, specifically the pooled NE Zambians (13%) and the Fwe and Shanjo of SW Zambia (24-27%).
In any case it is a notorious founder effect, almost absent in other Bantus of the area (0-10%).
Typical L0d Khoisan admixture:
This element is concentrated in Botswana (~25%) and with highest frequencies in the SW Kgalagadi (53%). It is also important among the Kuvale of SW Angola (21%). Other Bantu populations in this dataset have frequencies under 10%, some even zero. The Damara have 13%.
We know from previous studies that it is also found at high frequencies among the Xosha of South Africa (L0d3).
While L3h appears marked in the graph, the lineage is in fact absent in all populations except at very low frequency among the Kuvale (2%), so it does not seem actually of any relevance.
Less typical L0k around SW Zambia:
While L0k is generally considered an aboriginal Southern African lineage it has a much more northernly distribution than the more common and surely older L0d. Its area of greatest commonality seems to be SW Zambia (see
here and
here).
This study confirms this distribution:
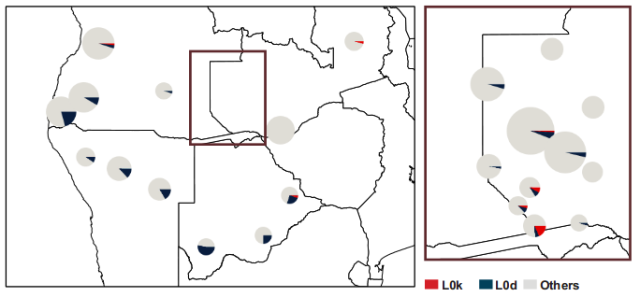 |
|
Supplementary Figure S3[A]: Haplogroup frequencies of important haplogroups in the populations studied here. A: Haplogroups L0d and L0k.(…)
The size of the circles is proportional to the sample size. |
High frequencies of L1c (Pygmy admixture marker) among Southern African Bantus:
An interesting element is the commonality of L1c,
typical of Western Pygmies and some other populations from Gabon (possibly representative of the wider West-Central Africa jungle region, not too well studied otherwise), among almost all Bantu populations in this dataset.
The exceptions are the Herero, Himba, Kgalagadi and Tswana (0%), as well as the NE Zambians (4%). All the rest have frequencies between 12% and 30%. Even the non-Bantu Damaras have 11% of it.
In my understanding this almost certainly implies a notable level of admixture with Western Pygmies of the Bantus from especially Angola and West Zambia. A phenomenon that may be widespread in Central-West Africa.
It is notable however that at least many of the populations with the highest likely Khoisan admixture (in its various forms, discussed in the previous sections) have the lesser frequencies of L1c (Pygmy admixture). So to a great extent these two aboriginal influences in Bantu mtDNA seem mutually exclusive and were probably produced after settlement rather than “on the march”.
This in turn arises some interesting questions about the ethnic geography of Africa before the Bantu expansion.
Update: I just noticed that Ethiohelix has parsed the haplogroups’ frequency into a very helpful chart → LINK.
See also:
-
-
-
- My latest reconstruction of human early expansion in Africa (within a larger entry) → LINK
-
-
-
-
-
-